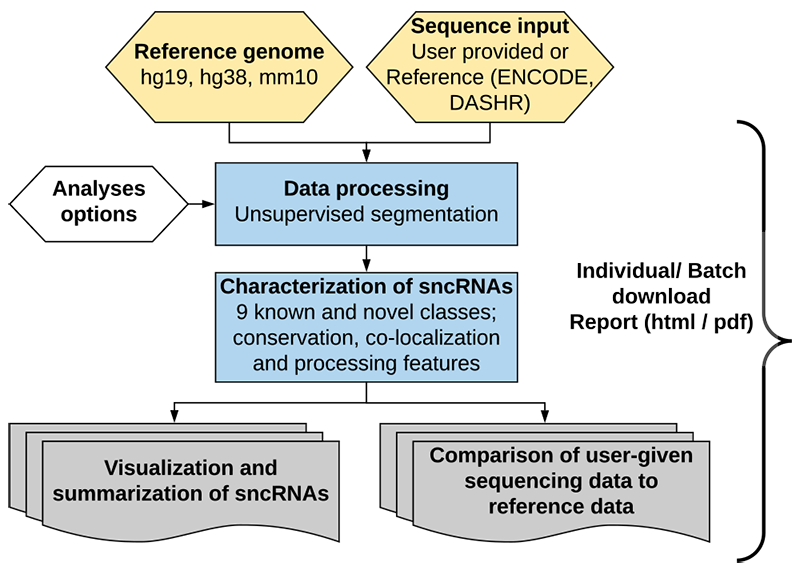
- Ab initio (annotation-free) small non-coding RNA (sncRNA) discovery and characterization
- Expression comparison of user data with >180 reference human tissues, cell types and cell lines from DASHR and ENCODE
- Annotations for GRCh37/hg19, GRCh38/hg38, mm10
Method
Distinctive key features:
Supported inputs and protocols:
- Small RNA-sequencing, microRNA-seq, short total RNA sequencing protocols
- Mapped reads (BAM), raw signal tracks (BigWig)
- Genome-wide or user-provided target region analysis
Deliverables:
- Visualization of small RNA-seq data: size and class composition, processing characteristics, genome-wide patterns
- Interactive sncRNA expression table for data exploration
- Direct Genome Browser view of individual loci
- Downloadable cross-reference comparison expression tables for identified sncRNA loci
- Downloadable signal and sncRNA locus segmentation tracks
SPAR integrates significantly improved mapping, segmentation, annotation, and RNA processing information for both sncRNA genes and mature sncRNA products (Leung et al. NAR 2016).
The improved mapping by SPAR provides significantly more data for downstream analysis (on average, over 28% more reads per library).
SPAR segmentation is not only signficantly more accurate, but also is order-of-magnitude faster (60x speed-up on average) by utilizing novel segmentation algorithms.
SPAR has been used to process over 800 smRNA high-throughput sequencing (smRNA-seq) datasets from GEO/SRA and ENCODE with over 20 billion reads.
SPAR results are integrated into the database of small human non-coding RNAs (DASHR, http://lisanwanglab.org/DASHR) described in NAR 2016 Database issue.
SPAR peak caller
The input to the algorithm are mapped reads from small RNA-seq experiment (BAM) or genome-wide read coverage profile (BigWig). The parameters are the minimum peak height (10 reads by default), and the mininum fold change in read depth (2 by default) for detecting peak starts.
The minimum peak width / read length (15) and the maximum peak width / read length (44) are other optional parameters that can be used to select the desired RNA length range for analysis.
The results of the algorithm are:
- Summary / QC report: lists the number of reads, the number of identified peaks (loci), the mean length of the peaks, the mean of the expression values, genome coverage (in nucleotides). These metrics are also shown separately for annotated and un-annotated loci.
- Peaks: the list of all called peaks. Also provided are separate lists for annotated, un-annotated peaks, as well separate lists for each class of RNA precursor. These are available in the BED6 format, as well as xls files with detailed information for each of the called peaks (such as conservation, genomic location relative to protein coding genes, cleaving/processing specificity, proportion of reads with the same 5'/3' end, etc)
- Tracks: genome-wide signal information in BigWig format. Separate tracks for called peaks, and raw read coverage are provided. The tracks can be loaded into UCSC Genome Browser or other genome browser for visualization / exploration. The peaks in the track files contain outlinks to DASHR database.
- Integrated Expression Tables: list called peaks along with their corresponding expression information in integrated reference datasets. Separated table are provided for reference datasets in DASHR and ENCODE.
SPAR pipeline
The SPAR pipeline involves the following steps:
- Call peaks corresponding to smRNA sites (ab initio annotation-free segmentation)
- Annotate called peaks by using integrated RNA annotation from DASHR
- Construct genome-wide tracks with called peaks and raw read coverage information
- Integrate called peaks with DASHR and ENCODE reference expression information
- Conservation, genomic location, sequence analyses
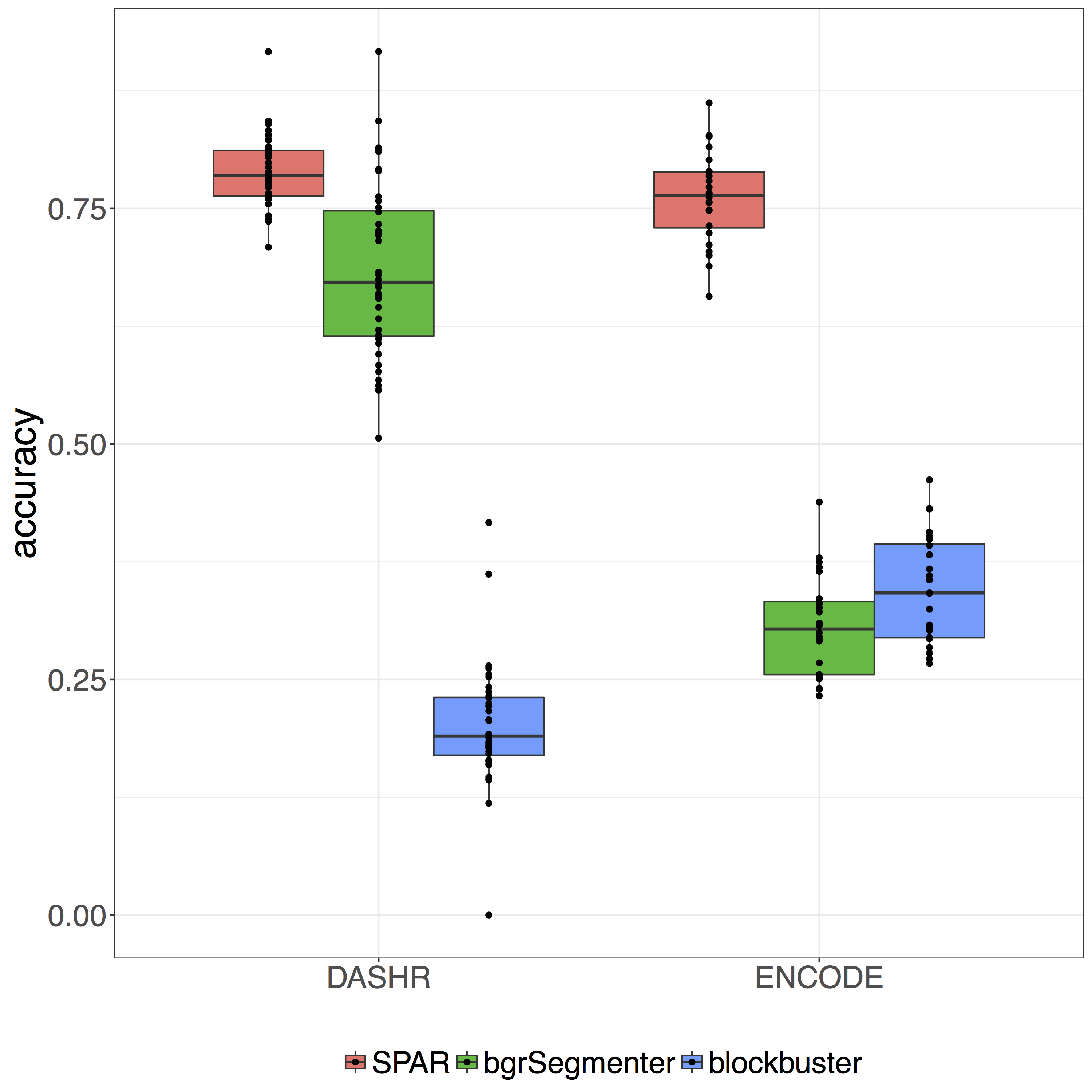
SPAR evaluation
We systematically evaluated the accuracy of SPAR in detecting smRNA sites using ENCODE and DASHR smRNA-seq datasets. To compute accuracy, we used miRNA loci from mirBase as the reference set. We also analyzed the accuracy of the baseline peak detectors on the same data.
Download
If you prefer to run SPAR at your own site, please download stand-alone, offline version here: SPAR repository
Citation
Pavel P. Kuksa, Alexandre Amlie-Wolf, Zivadin Katanic, Otto Valladares, Li-San Wang, Yuk Yee Leung. SPAR: small RNA-seq portal for analysis of sequencing experiments. NAR 2018 (Web Server issue). https://doi.org/10.1093/nar/gky330